News
August 9, 2022
Clearing the mist hiding the genome
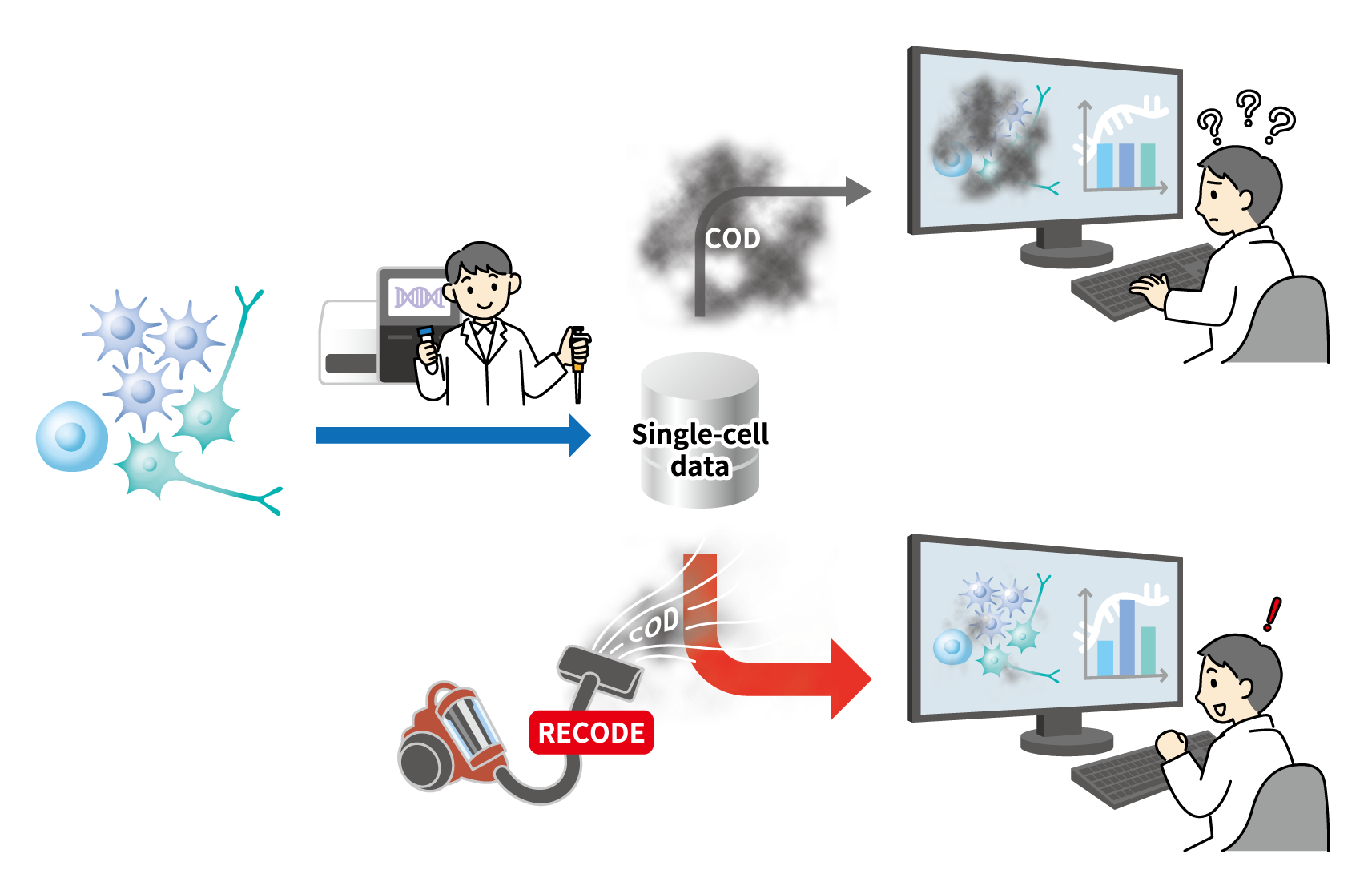
Graphical abstract. (Credits: TRAIS )
Since scientists first mapped the complete human genome, attention has now turned to the question of how cells use this master copy of genetic instructions. It is known that when genes are switched on, parts of the DNA sequences in the cell nucleus are copied into shorter string-like molecules, RNA, which deliver the molecules essential for survival and cell-specific functions.
Understanding the profiles of RNAs in a cell can show which genes are active and allow researchers to speculate what the cell is doing. The technology for measuring RNA by massively parallel DNA sequencer, RNA-sequencing, has become a standard technique over the past decade. More recently, rapid technological advances permit RNA sequencing at the single-cell level from thousands of cells in parallel, accelerating progress in the biomedical sciences. But quantifying RNAs from such a tiny material poses great technical challenges. Even with state-of-the-art equipment, data produced from single-cell RNA sequencing data contain large detection errors, including the so-called “drop-out effect”. Moreover, even small errors in the calculations for a large number of genes can quickly add up so that any useful information is lost among signal noise.
Now, a team from the Kyoto University Institute for Advanced Study of Human Biology (WPI-ASHBi) has developed a new mathematical method that can eliminate the noise and thus enable the extraction of clear signals from single-cell RNA sequencing data. The new method successfully decreases random sampling noise in the data to enable a precise and complete understanding of a cell’s activity. The research has recently been published in the journal Life Science Alliance.
The lead author of the paper, Yusuke Imoto from ASHBi, explains, “Each gene represents a different dimension in RNA sequencing data, which means that tens of thousands of dimensions must be collected across multiple cells and analyzed. Even the slightest noise in one dimension can majorly impact the downstream data analyses so that potentially important signals are lost. This is why we call this the “curse of dimensionality.”
To break the curse of dimensionality, the Kyoto team has developed a new noise reduction method, RECODE—standing for “resolution of the curse of dimensionality”—to remove the random sampling noise from single-cell RNA sequencing data. RECODE applies high-dimensional statistical theories to recover accurate results, even for genes expressed at very low levels.
First, the team tested their method on data from a broadly well-studied cell population, human peripheral blood. They confirmed that RECODE successfully removes the curse of dimensionality to reveal expression patterns for individual genes close to their expected values.
Next, when compared against other state-of-the-art analysis methods, RECODE outperformed the competition by giving much truer representations of gene activation. Moreover, RECODE is simpler to use than other methods, without relying on parameters or using machine learning for the calculations to work.
Finally, the team tested RECODE on a complex dataset from mouse embryo cells containing many different types of cells with unique gene expression patterns. Whereas other methods blurred the results, RECODE clearly resolved gene expression levels, even for rare cell types.
Imoto concludes, “Single-cell RNA sequencing data analysis remains technically challenging and is a developing technique, but our RECODE algorithm is a step towards being able to reveal the true behaviors of single-cell structures. With our contribution, single-cell RNA sequencing data analysis could become a powerful research tool with massive implications across many biological fields.” Another leading author Tomonori Nakamura, a biologist from ASHBi and The Hakubi Center for Advanced Study, Kyoto University, adds, “By unlocking the true power of single-cell RNA sequencing, RECODE will enable researchers to discover unidentified rare cell types, leading to the development and establishment of the new research field in basic science as well as clinical application and drug discovery research.”
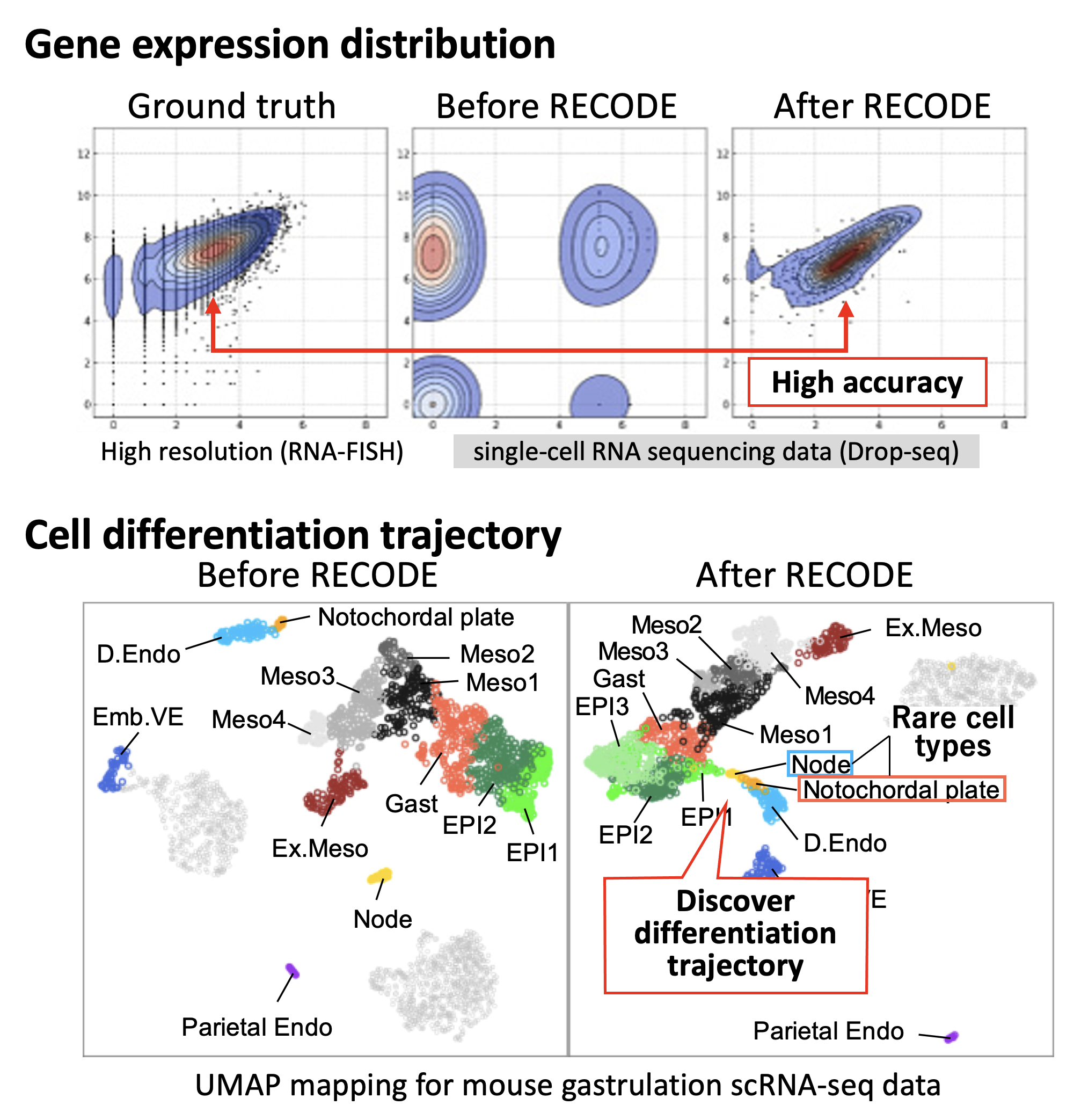
Comparison of RECODE performance. (Top) Recovery of gene expression distribution (Bottom) Detection of rare cell type (Node・Notochordal plate) and discovery of cell differentiation trajectory.
Paper Information
Yusuke Imoto, Tomonori Nakamura, Emerson G. Escolar, Michio Yoshiwaki, Yoji Kojima, Yukihiro Yabuta, Yoshitaka Katou, Takuya Yamamoto, Yasuaki Hiraoka, Mitinori Saitou. (2022) “Resolution of the curse of dimensionality in single-cell RNA sequencing data analysis.” Life Science Alliance, DOI: https://doi.org/10.26508/lsa.202201591
Reference
RECODE calculation programs (Python/R code, desktop application) are available on GitHub (https://github.com/yusuke-imoto-lab/RECODE).