News
June 24, 2020
Identification of distinct loci for de novo DNA methylation
DNA methylation (*1) performs an essential function in mammalian ontogeny. It is also known that abnormalities in this process cause the development of cancers such as leukemia. A research team working at The University of Tokyo and Kyoto University in Japan has announced that they have successfully identified specific target sites for the DNA methylases DNMT3A and DNMT3B (*2 ).
The researchers also found that DNMT3A specifically regulates differentiation-related genes and DNMT3B specifically regulates X-chromosomal genes during mammalian ontogeny. These results were published today in Nature Communications (online version).
“The results will lead to the elucidation of the regulatory mechanism of DNA methylation during the mammalian developmental process and also lead to a more detailed understanding of the onset mechanism of cancer and other diseases caused by abnormal DNA methylation.,” said the principal investigator of this research, Professor Yasuhiro Yamada at the Division of Stem Cell Pathology, Center for Experimental Medicine and Systems Biology, The Institute of Medical Science, The University of Tokyo (IMSUT).
Two conserved de novo DNA methyltransferases, DNMT3A and DNMT3B
DNA methylation plays essential roles in mammalian development. Two conserved DNA methyltransferases, DNMT3A and DNMT3B are responsible for de novo establishment of DNA methylation patterns during mammalian development.
Somatic inactivating mutations in DNMT3A and DNMT3B have been reported in diseases such as hematologic cancers and immune disorders, suggesting that DNMT3A and DNMT3B are essential for proper maintenance of somatic cells. At present, the distinct functions of these two enzymes remain largely elusive.
To comprehensively identify the target sites for de novo DNA methylation by the DNMT3 enzymes, the research team took advantage of female mouse ES cells that lack most DNA methylation (2i/L ES cells) (*3), in combination with genetic ablation of Dnmt3a or Dnmt3b (Figure 1). They analyzed de novo DNA methylation in mouse embryonic fibroblasts (2i-MEFs) derived from Dnmt3 knockout (KO) 2i/L ES cells.
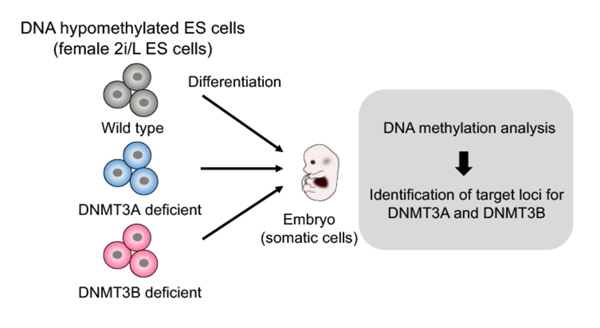
Figure 1. Schematic of experimental design
Both Dnmt3a and Dnmt3b KO 2i-MEFs exhibited a modest but global reduction in DNA methylation, which was particularly notable on the X chromosome in Dnmt3b KO cells. Although most genes were methylated similarly in both knockouts, they identified 355 and 333 uniquely unmethylated genes in Dnmt3a and Dnmt3b KO 2i-MEFs, respectively.
Notably, Dnmt3a was exclusively required for de novo methylation at both TSS regions and gene bodies of Polycomb group (PcG) target developmental genes. Consistent with this, tissue-specific DNA methylation at PcG target developmental genes was substantially reduced in Dnmt3a KO embryos (Figure 2).
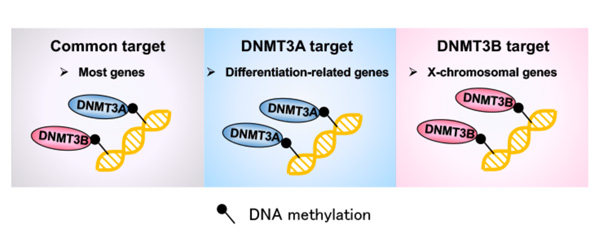
Figure 2. Summary of this study
Finally, the research team found that human patients with DNMT3A mutant acute myeloid leukemia (AML) or harboring DNMT3B mutations associated with immunodeficiency, centromere, and facial anomalies (ICF) syndrome exhibited reduced DNA methylation at regions that were hypomethylated in Dnmt3a or Dnmt3b KO 2i-MEFs, respectively.
Potential impact of the research
The researchers reported a set of unique de novo DNA methylation target sites for both DNMT3 enzymes during mammalian development that overlap with hypomethylated sites in human patients.
Professor Yamada states the work’s importance as a potential for further; ” For the first time in the world, we have succeeded in identifying the target genes of DNMT3A and DNMT3B during the mammalian developmental process, which up till now were unknown. Our findings may contribute to the development of a novel therapeutic approach for patients with DNMT3 mutations.”
This study was supported in part by P-CREATE (JP18cm0106203h0003), Japan Agency for Medical Research and Development (AMED); AMED-CREST (JP19gm1110004h9903), AMED; JSPS KAKENHI (18H04026, 20H05384); the Princess Takamatsu Cancer Research Fund; the Takeda Science Foundation; the Mochida Foundation; and the Naito Foundation to Y. Y.. T. Y. was supported by AMED-CREST (19gm1110004s0203); JSPS KAKENHI 19H03418; Core Center for iPS Cell Research, Research Center Network for Realization of Regenerative Medicine, AMED; and the iPS Cell Research Fund. M. Y. was supported by JSPS KAKENHI 15J05792. The ASHBi is supported by World Premier International Research Center Initiative (WPI), MEXT, Japan.
Research Notes
- (*1) DNA methylation
A biological process by which methyl groups are added to the DNA molecule, which is important for regulation of gene expression. - (*2) DNA methylases DNMT3A and DNMT3B
DNMT3A and DNMT3B, encoded by the Dnmt3a and Dnmt3b genes, respectively, function in establishing DNA methylation patterns during embryogenesis. Mutations at Dnmt3a and Dnmt3b cause various diseases, including cancers and immune disorders. - (*3) 2i/L ES cells
Embryonic stem (ES) cells established in the presence of two chemical compounds, MEK and GSK3 inhibitors. Female 2i/L ES cells lack most DNA methylation while the conventional ES cells harbor higher levels of DNA methylation.
Reference
Yagi M, Kishigami S, Tanaka A, Semi K, Mizutani E, Wakayama S, Wakayama T, *Yamamoto T, *Yamada Y. Derivation of ground-state female ES cells maintaining gamete-derived DNA methylation. Nature. 2017 Aug 10;548(7666):224-227.Paper Information
- Journal: Nature Communications
- Title: Identification of distinct loci for de novo DNA methylation by DNMT3A and DNMT3B during mammalian development
- Authors: Masaki Yagi#, Mio Kabata#, Akito Tanaka, Tomoyo Ukai, Sho Ohta, Kazuhiko Nakabayashi, Masahito Shimizu, Kenichiro Hata, Alexander Meissner, Takuya Yamamoto*, Yasuhiro Yamada*
#These authors contributed equally
*Corresponding author
- Masaki Yagi#
Department of Molecular Biology, Massachusetts General Hospital
At the time of research:Division of Stem Cell Pathology, Center for Experimental Medicine and Systems Biology, The Institute of Medical Science, The University of Tokyo - Mio Kabata#
Department of Life Science Frontiers, Center for iPS Cell Research and Application (CiRA), Kyoto University - Takuya Yamamoto*
Department of Life Science Frontiers, Center for iPS Cell Research and Application (CiRA), Kyoto University
Institute for the Advanced Study of Human Biology (WPI-ASHBi), Kyoto University
Medical-risk Avoidance based on iPS Cells Team, RIKEN Center for Advanced Intelligence Project (AIP) - Yasuhiro Yamada*
Division of Stem Cell Pathology, Center for Experimental Medicine and Systems Biology, The Institute of Medical Science, The University of Tokyo
- Masaki Yagi#
- doi: https://doi.org/10.1038/s41467-020-16989-w
Source: CiRA