
Guillaume Bourque
PI
- Position
- 特別招へい教授
- Research Field
- Bioinformatics, Genomics, Epigenomics
- Personal Website
- http://www.computationalgenomics.ca/BourqueLab/
研究概要
We seek to understand transcription regulation in mammals and characterize the contribution of non-coding
DNA variants to human disease. With consortiums such as ENCODE and the International Human Epigenome
Consortium (IHEC), the number of datasets characterizing the epigenomic state of different cell types and
conditions has been steadily growing. Nonetheless, studies trying to infer the impact of non-coding DNA
variants have had to rely on limited functional datasets to train their model and make predictions, which
has proven difficult. We have shown that by using an evolutionary perspective to interpret functional
genomic datasets, we can better decrypt the role of non-coding human DNA. To improve on this even further,
we now propose to build comprehensive epigenomic profiles of specific cell-types in multiple human
individuals and in various non-human primate species (including chimpanzee, bonobo, gorilla, rhesus and
marmoset). We will then use these datasets profiling intra and inter-species epigenomic variation, to
develop and train algorithms to predict the impact of non-coding DNA variants in unprecedented ways
(Figure 1).
Areas of interest include: the evolution of regulatory sequences, the role of transposable elements in
gene regulation and the impact of genome rearrangements in evolution and cancer. One objective is to
develop computational methods and resources for the functional annotation of genomes with a special
emphasis on sequencing-based assays (e.g. ChIP-seq, RNA-Seq, exome- and whole-genome sequencing,
single-cell analysis).
We are also involved in the Global Alliance for Genomics and Health (GA4GH) and develop tools which
facilitates data sharing(Figure 2).
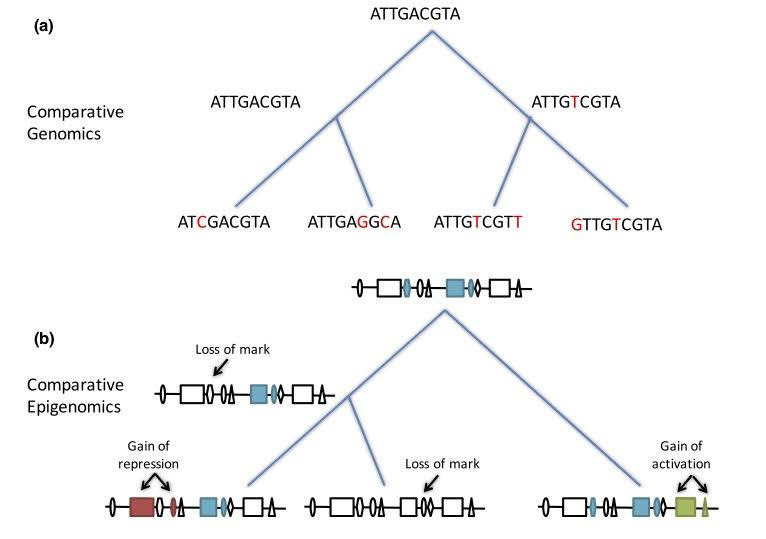
Figure 1. (a) Comparative genomics ancestral genome reconstruction using phylogenetic tree (maximum
parsimony method). (b) Ancestral epigenome reconstruction using parsimony method and counting loss,
change in state, and adding of epigenomic marks as mutations (from Venuto and Bourque 2018).
Figure 2. Screenshot of the International Human Epigenome Consortium (IHEC) Data Portal (Bujold et al.
2016) a tool we developed to facilitate analysis and sharing of epigenomic datasets.
論文
Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvak Z, Levin HL, Macfarlan TS, Mager DL & Feschotte C. Ten things you should know about transposable elements. Genome Biol 2018. 19(1):199.
Goerner-Potvin P. and Bourque G. Computational tools to unmask transposable elements. Nat Rev Genet 2018. 19, 688-704.
Venuto D, Bourque G. Identifying co-opted transposable elements using comparative epigenomics. Dev Growth Differ. 2018. 60(1):53-62.
Monlong J, Cossette P, Meloche C, Rouleau G, Girard SL, Bourque G. Human copy number variants are enriched in regions of low-mappability. NAR 2018. 46(14):7236-7249.
Bujold D, Morais D, Gauthier C, Côté C, Caron M, Kwan T, Chen KC, Laperle J, Markovits AN, Pastinen T, Caron B, Veilleux A, Jacques PE, Bourque G. IHEC Data Portal: a resource for discovering, analysing and sharing epigenomics data. Cell Syst. 2016 3(5):496-499.e2.